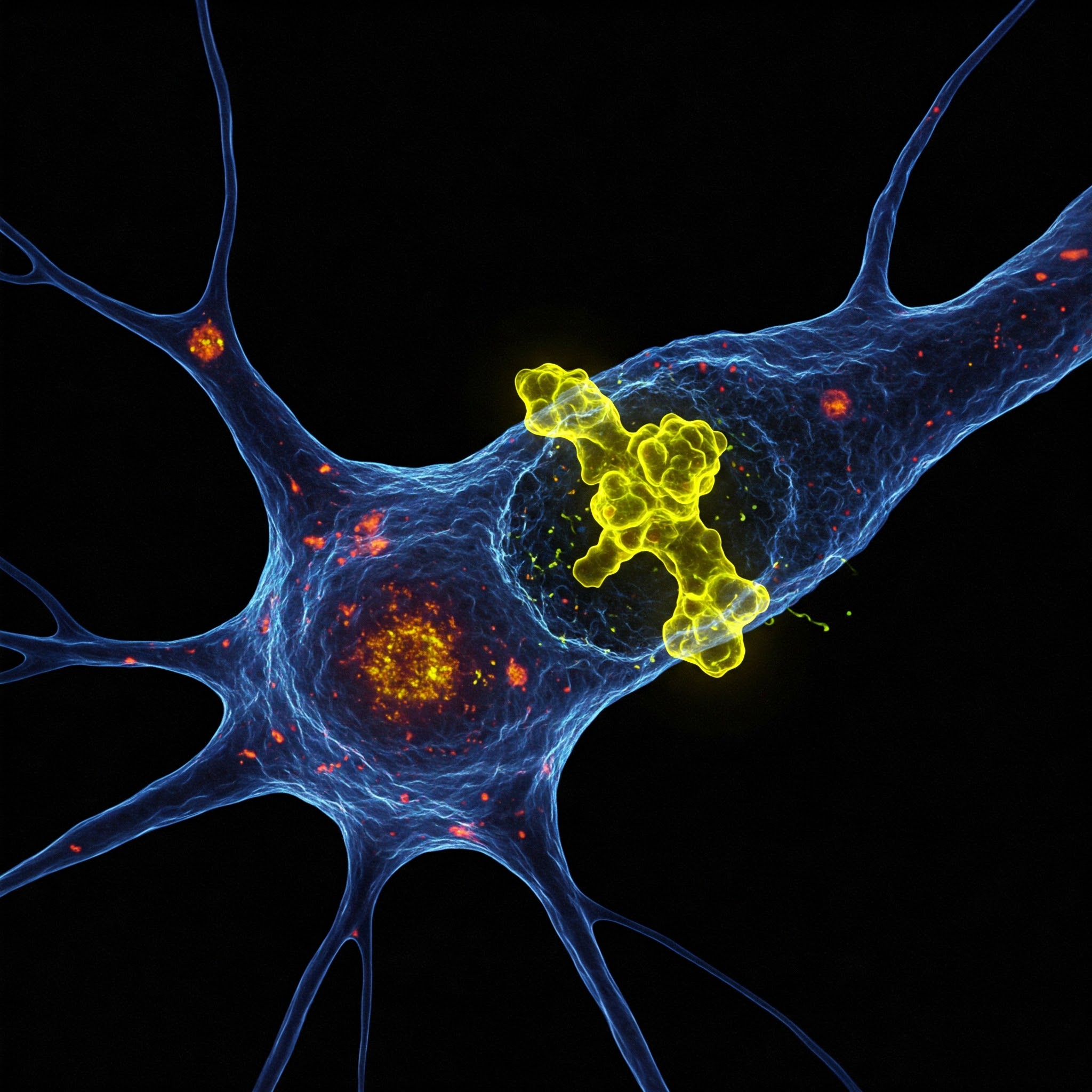
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are devastating neurodegenerative diseases with no cure. While both conditions exhibit distinct symptoms, recent research suggests they share common pathological mechanisms, particularly involving the misfolding and aggregation of the TDP-43 protein. In a groundbreaking study, Belgian scientists have successfully engineered lab-made protein fragments that replicate these harmful processes in human cells. This discovery sheds light on how these diseases progress and offers a promising avenue for developing targeted therapies.
The Role of TDP-43 in ALS and FTD
TAR DNA-binding protein 43 (TDP-43) is a crucial regulator of RNA metabolism and cellular function. In healthy cells, TDP-43 resides primarily in the nucleus, where it facilitates gene expression and protein synthesis. However, in ALS and FTD, TDP-43 becomes mislocalized to the cytoplasm, where it forms toxic aggregates. This disruption triggers a cascade of cellular dysfunctions, ultimately leading to neurodegeneration.
The accumulation of TDP-43 in brain cells is now considered a hallmark of ALS and FTD, appearing in nearly 97% of ALS cases and about 50% of FTD cases. Understanding how and why this mislocalization occurs has been a critical challenge in neuroscience.
Breakthrough: Lab-Made Protein Fragments Mimic Disease Progression
A team of Belgian researchers has successfully synthesized protein fragments that mimic the behavior of TDP-43 in ALS and FTD. By engineering these protein fragments in the lab, scientists were able to induce mislocalization and aggregation of TDP-43 in human cells. This groundbreaking achievement provides a new experimental model to study the pathology of these diseases in greater detail.
How the Experiment Works
The researchers created small protein fragments derived from TDP-43 and introduced them into cultured human cells. They observed the following key processes:
- Mislocalization – The engineered protein fragments caused TDP-43 to relocate from the nucleus to the cytoplasm.
- Aggregation – Once in the cytoplasm, TDP-43 formed toxic clumps similar to those seen in ALS and FTD patients.
- Cellular Dysfunction – The accumulation of these protein aggregates led to impaired cellular functions, including RNA processing errors and increased cell stress.
These findings confirm the prion-like behavior of TDP-43, where misfolded proteins trigger further misfolding, potentially explaining how ALS and FTD spread within the brain.
The “Prion-Like” Nature of ALS and FTD
The idea that ALS and FTD progress in a prion-like manner has gained traction in recent years. Prion diseases, such as Creutzfeldt-Jakob disease, involve misfolded proteins that act as templates, causing other normal proteins to misfold and aggregate. Similarly, misfolded TDP-43 appears to propagate its toxic effects by recruiting healthy proteins into harmful aggregates.
This new research supports the prion-like hypothesis, as the introduction of lab-made protein fragments into human cells effectively initiated the same disease processes observed in ALS and FTD patients.
Implications for ALS and FTD Research
The ability to replicate ALS and FTD pathology in human cells using lab-made protein fragments opens up several promising research avenues:
1. Developing Better Disease Models
Traditional animal and cell models have struggled to fully replicate the complex pathology of ALS and FTD. This new approach provides a more accurate system for studying disease mechanisms at the molecular level.
2. Identifying Potential Drug Targets
By understanding how TDP-43 mislocalization and aggregation occur, researchers can identify specific molecular targets for therapeutic intervention. Drugs that prevent or reverse this mislocalization could slow or stop disease progression.
3. Screening for New Treatments
The lab-made protein fragments create a standardized model for testing potential treatments. Pharmaceutical companies can use this system to evaluate the efficacy of drugs designed to reduce TDP-43 aggregation or enhance its clearance from the cytoplasm.
Challenges and Future Directions
Despite this exciting breakthrough, several challenges remain:
- Translating Lab Findings to Human Treatment: While these protein fragments work in cultured cells, it remains to be seen whether they will behave similarly in live human neurons.
- Understanding Disease Onset: It is still unclear what triggers TDP-43 mislocalization in the first place. Uncovering these initial steps is crucial for early intervention.
- Ensuring Safety of Therapeutic Approaches: Any treatment targeting TDP-43 must preserve its essential functions while preventing aggregation.
Conclusion
The creation of lab-made protein fragments that mimic ALS and FTD pathology marks a significant step forward in neurodegenerative disease research. By replicating disease processes in human cells, scientists can now explore novel treatment strategies with greater precision. This discovery not only enhances our understanding of how ALS and FTD progress but also paves the way for new therapies that could ultimately improve the lives of those affected by these devastating conditions.
As researchers continue to build on this foundation, the future of ALS and FTD treatment looks increasingly promising. The hope is that by targeting the root causes of TDP-43 dysfunction, we can move closer to a cure for these relentless diseases.
Also Read:
New technique can detect TDP-43 protein in patients with ALS and frontotemporal dementia







+ There are no comments
Add yours